Hlavní menu
Sociální sítě:



Souhrnný článek představující projekt Quantum Monte Carlo at Home. Jedná se o projekt snažící se prostřednictvím kvantové chemie a systému Boinc získat znalosti o molekulárních strukturách a reaktivitě molekul.
Žijeme ve světě plném molekul. Z molekul se skládá i naše tělo a reakce mezi molekulami jsou zásadně důležité pro všechny životní procesy. Dýcháme, pojídáme a oblékáme si molekuly každý den. Pokud si toto uvědomíme, můžeme si představit důležitost znalostí o molekulárních strukturách a také užitečnost schopnosti přesně předpovídat reaktivitu molekul.
Kvantový svět je jiný než je náš "velký" svět, protože popisuje hmotu a její chování na její nejzákladnější (dosud známé) úrovni. Náš "velký" (makro)svět je už jen statistickým příměrem kvantového (mikro) světa. Proto jsou v kvantové mechanice možné různé věci, které nám připadají absurdní, - např. procházení částic přes potenciálové bariéry (stěny), existence částice ve dvou stavech současně (zároveň jsou i nejsou), průchod elektronů přes dva otvory současně a podobně. Kvantová mechanika je ohromně obsáhlá věda, ale není konkrétním předmětem výzkumu na tomto projektu, je pouze jeho nástrojem.
Kvantová teorie nám v principu umožňuje předpovídat strukturu a reaktivitu všech molekul, ovšem s rostoucí velikostí systému (molekuly) se stávají rovnice kvantové teorie příliš složité. Přesné analytické řešení je možné jen pro nejmenší systémy, ale pro téměř všechny zajímavé molekuly v chemii a „life sciences“ (vědách o životě) taková řešení neznáme.
Kvantová chemie je větev teoretické chemie,která používá kvantové mechanismy a kvantovou teorii pro řešení úloh v chemii. Popis chování atomů v molekule ve vztahu k jejich reaktivitě je jeden ze základů kvantové chemie. Kvantová chemie leží na hranici mezi chemií a fyzikou a významně k ní přispívají vědci z obou oborů. Silně se jí například dotýká oblast atomové fyziky a molekulární fyziky, stejně jako fyzikální chemie a chemické fyziky.
Kvantová mechanika nahradila newtonskou mechaniku a klasický elektromagnetismus, které nedokázaly vysvětlit pozorování na atomových a vnitroatomárních úrovních. Kvantově mechanické simulace nejsou založeny na zákonech klasické newtonovské mechaniky, ale využívají zákonů kvantové mechaniky.
Historie kvantové chemie začala v podstatě v roce 1838 objevem katodových paprsků Michaelem Faradayem a dále pokračovala kolem roku 1859 výzkumem Gustava Kirchhoffa a okolo 1877 návrhy od Ludwiga Boltzmanna. V roce 1900 přichází s kvantovou hypotézou Max Planck. Tato hypotéza předpokládá, že každý energetický systém atomů může být rozdělen do několika menších energetických částí tak, že každá z těchto částí je úměrná kmitočtu, který každá jednotlivě vyzařují.
V roce 1926 byla formulována Schrödingerova rovnice. Dva roky poté publikoval Hylleraas kvantově mechanické výpočty pro atom He. V roce 1930 D. Hartree a V. Fock vypracovali metodu self-konsistentního pole (SCF). Tato metoda se také nazývá Hartree-Fockova metoda a zavádí aproximace, usnadňující řešení Schrödingerovy rovnice. Krátce nato byla uvedená metoda použita pro kvantově mechanické výpočty molekuly H2 (James a Coolidge, 1933). Vývoj na poli kvantové mechaniky utlumila druhá světová válka. Až v roce 1950 se objevily další práce v této oblasti: Například C. C. Roothan publikoval maticovou verzi Hartree-Fockovy metody (ta se používá s malými úpravami až dosud).
Od 50. let se kvantová mechanika rozděluje na dva směry: metody semiempirické a ab initio. Jako první dostal za kvantovou chemii Nobelovu cenu Linus Pauling v roce 1954.
Schrödingerova rovnice je jedna ze základních rovnic kvantové mechaniky a kdysi si ji geniální Schrödinger na základě logiky a intuice doslovně "vycucal z prstu". Až později byla tato rovnice dodatečně odvozena.
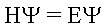 E - energetický stav molekuly
E - energetický stav molekuly
H - Hamiltonův operátor
Ψ - vlnová funkce (definována strukturou molekuly)
Ani pro velmi malé molekuly není možno získat se současnými výpočetními prostředky přesné řešení Schrödingerovy rovnice. Proto obsahují kvantově mechanické metody mnoho různých matematických aproximací (přibližných hodnot).
Velmi laicky lze říci, že tato rovnice nám říká, kde v prostoru se s jakou pravděpodobností částice právě nachází a jak se tato pravděpodobnost vyvíjí v čase. Částice jsou v ní popisované jako "oblaky pravděpodobnosti", jako jakési rozmazané chomáče, které víří v prostoru jako včelí roj. Taková je totiž přirozenost našeho světa na kvantové úrovni. A tak ji tedy i Schrödingerova rovnice popisuje. Rovnice to není složitá (pokud ovládáte vyšší matematiku), ale její řešení je velmi složité. Řešení je možné jen v některých specifických případech a vždy se jedná jen o numerické přiblížení, kde nám musí pomoci mnohé finty matematiky (kterých je naštěstí poměrně hodně a jsou častokrát velmi účinné).
Kvantová chemie je věda, která vymýšlí aproximace rovnic kvantové teorie k předpovídání molekulárních informací s velkou přesností. Avšak i řešení těchto jen aproximovaných kvantově chemických rovnic v systémech reálného života vyžaduje obrovské množství výpočetního výkonu a objemu výpočtů.
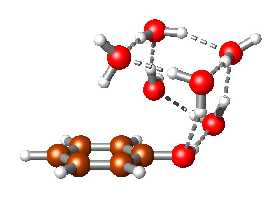
Moderní kvantověmechanické metody slouží k interpretaci mnohých experimentálních výsledků a umožňují modelování dějů v různých oborech chemie i fyziky. Mohou být používány při studiu velkých biologických systémů, jakým je například životně důležitá dezoxyribonukleonová kyselina (DNA), i při modelování technologicky významných katalytických procesů. V zájmu osvětlení funkce DNA byly studovány interakce dusíkatých zásad, které jsou její nejdůležitější složkou.
Za projektem stojí Univerzita v Münsteru (Německo). Projekt QMC@Home se zabývá aplikací kvantové mechaniky ve světě molekul - tedy ne na úrovni atomů a částic, jak to dělá fyzika. Kromě této rozměrové škály jinak v podstatě není mezi kvantovou chemií a fyzikou žádný rozdíl. Problém při kvantověmechanickém popisu systémů skládajících se z více atomů je ten, že je to ohromně výpočetně náročný úkol. Náročnost výpočtů roste zhruba s třetí mocninou velikosti systému, a teda když máte desetkrát větší systém, potřebujete tisíckrát delší čas (či větší výkon) na výpočet jeho vlastností. Proto se s kvantověmechanickými výpočty (ab initio výpočty) nedostaneme daleko. Například Juraj Kotulič Bunta (Duro) z Japan Atomic Energy Agency o těchto výpočtech říká: ,,Já osobně jsem se ani s nejnovějšími japonskými superpočítači nedostal dále než k molekule o počtu atomů 112, a jiní kolegové se nedostali o moc dále,, . To jsou v biochemii velmi malé molekuly a dá se takto počítat tedy jen velmi úzký okruh. Jaké je řešení?
Jedna z nových a perspektivních řešení se ukazuje být stará známá metoda Monte Carlo simulace. Dávno se používají v jiných oblastech (např. v jaderné fyzice na simulace interakce částic v urychlovačích s bombardovanými terči).

Tým lidí starajících se o provoz projektu QMC@Home.
V zásadě se dá říci, že při hledání řešení se využívají náhodná čísla (proto název "Monte Carlo", (i když pro přesnost je třeba uvést, že se nejedná o úplně náhodná čísla). U metody Monte Carlo jde o výpočty, u kterých jsou vstupní data rozdělena na dvě části. První tvoří soubor pevně daných parametrů (například rovnice, definice velikosti či prostoru) pro výpočet a jeho průběh a druhou část tvoří parametry nastavitelné (například koncentrace určité látky, vzorce atd.). Některé z nastavitelných parametrů se mohou stát pro některou sérii modelů i pevnými, aby se tak omezilo množství možných modelů, ale to už si řídí každý projekt zvlášť. Metodou Monte Carlo tedy vzniká velká spousta modelů (většinou nahodile) a tyto modely se po zpracování porovnávají, analyzují a vyhodnocují.
Ve vztahu k projektu QMC si metodu Monte Carlo můžeme představit jako vícerozměrný prostor (o daném počtu proměnných), kde se zvolí závislá proměnná, (obvykle minimální energie), nastaví se rozmezí proměnných hodnot a můžeme začít s výpočty.
Tak jako v jaderné fyzice "střílíte" velké množství náhodných částic do simulovaného terče a výsledkem takové simulace je pěkný graf rozdělení částic podle různých kritérií, tak i Schrödingerova rovnice se dá řešit "střílením" množství čísel a postupným nacházením řešení. Je to však ( podobně jako jiné přístupy) výpočtově velmi náročná úloha. Dá se u ní ovšem využít její další velký plus a tím je rozdělit na velký počet částečných výpočtů, jejichž výsledky se dají složit! A už jistě víte, v čem je síla této metody a proč se právě tato (a ne jiná kvantověmechanická metoda) objevila pod platformou Boinc. Váš počítač bude zpracovávat malou část z ohromného spektra výpočtů potřebných pro nalezení řešení Schrödingerovy rovnice.
Po složení množství výsledků se může dospět jednak ke konkrétním výsledkům pro samotné molekuly (jejich strukturu a interakci s jinými molekulami) a stejně tak i ke zdokonalení samotné kvantové Monte Carlo metody.
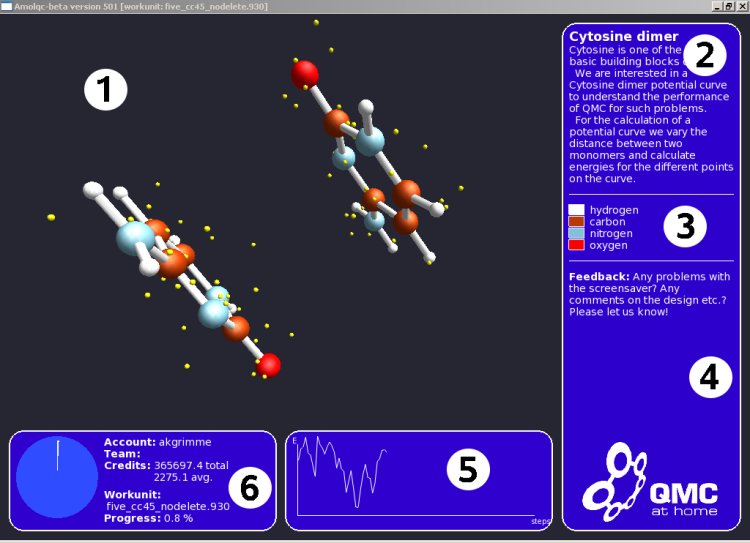
1. Vlastní interaktivní obraz molekuly.
2. Jméno molekuly a informace o tom proč je pro projekt zajímavý.
3. Legenda popisující části modelu.
4. Zprávy, tipy a informace.
5. Zobrazení křivky energie.
6. Jméno účtu a pracovních dat.
Popis toho, co je konkrétně na tomto screenshotu:
Základní stavební částí lidské DNA je cytosin (jeden ze čtyř nukleotidů, ze kterých se skládá DNA). Pro vědce je extrémně důležité přesně vypočítat jak cytosin reaguje s jinými molekulami, protože jen tak se dá zjistit, jak reagují různé proteiny (a tedy i léky) s naší DNA a mohou tak např. opravovat poškození DNA (rakovina, atd). Aby mohli autoři projektu vylepšit kvantovou Monte Carlo metodu kterou k výpočtům používají, potřebují začít na jednodušších úlohách. Na tomto konkrétním screenshotu jsou tedy dva cytosiny, pro které se počítá jejich vzájemná interakce - ve formě potenciální energie (něco jako elektrostatické napětí - přitahování či odpuzování), a to pro různé pozice a vzdálenosti těchto dvou molekul. Vizualizace (1) je velmi zjednodušená - kuličky jsou atomy, malé poletující tečky jsou elektrony, a paličky jsou vazby mezi atomy. Ve skutečnosti je to mnohem více kvantové, to znamená že se jedná spíše o obláčky elektronů a atomových jader. Takto zobrazené je to ale efektnější a přehlednější.
Cílem je tedy vypočítat celkovou energii sestavy, která se zobrazuje dole jako nepravidelná křivka (5). Když je křivka vysoko, znamená to, že v dané pozici se obě molekuly silně přitahují. Když je nízko, molekuly se přitahují málo nebo se dokonce odpuzují, v závislosti na různé pozici a vzdálenosti zúčastněných molekul. Díky tomu dokáží vědci zjistit, zda je algoritmus výpočtu dobrý a zda metoda funguje. Interakce dvou cytosinů je totiž jedna z nejlépe prozkoumaných a i experimentálně známých interakcí biomolekul. Když tyto křivky nebudou souhlasit se známými hodnotami, metodu bude třeba vylepšit.
Tuto testovací fázi, která probíhala pomocí tří po sobě jdoucích sérií WU Cytosine dimer má již projekt úspěšně za sebou a tak došlo počátkem května 2007 k ostrému spuštění a distribuování práce, pro kterou byl projekt vytvořen.
![]()
Jednotky QMC@Home (WU) jsou poměrně náročné na čas i paměť počítače které souvisejí s velikostí zpracovávaných systémů. Nyní (6/2007) zaberou výpočty projektu 4 - 48 hodiny na jednojádrovém 2,4 GHz systému. Cílem do budoucna je zdokonalit algoritmy a zmenšit WU tak, aby zpracovávání WU netrvalo déle než 3 dny na 2400 MHz systému a to i při těch nejnáročnějších modelech, které se postupně na projektu budou zpracovávat. Dále by počítač měl mít minimálně 512MB operační paměti.
Vstupní soubory se také budou časem zvětšovat. I v tomto ohledu je ovšem plánováno omezení na maximálních 10MB/WU. Termín odevzdání stahované práce je 2 týdny, což je úměrné času nutnému pro zpracování a náročnosti výpočtů. V průběhu výpočtů dochází k ukládání již zpracované práce (checkpointy). Čas nastavený pro toto ukládání by se měl pohybovat v rozmezí každých 5- 20 minut.
Odhadovaný čas dokončení WU je skutečně pouze odhad. Tento údaj se neustále vašimi výpočty bude aktualizovat a upřesňovat, ovšem vzhledem k velmi rozmanité náročnosti WU na zpracování se může u jednotlivých WU hodně lišit. Nikdy by ale neměla délka výpočtu jedné jednotky přesáhnout uváděné 3 dny na porovnatelném počítači. Podle rozdílu ve výkonu vašeho počítače oproti vzorovému se ovšem tato maximální délka výpočtu může podstatně snížit, ale i zvýšit.

Ukázka certifikátu uživatele. Takovýto certifikát si může nechat vygenerovat každý počtář projektu QMC.
S ohledem na několik stížností v první testovací fázi projektu, kdy byl použit standardní BOINC kreditový mechanismus závislý na Banchmarku integrovaném přímo v systému BOINC, se vedení projektu rozhodlo od něj ustoupit. Nyní je kreditové ohodnocení vaší práce prováděno tak, že je každé sérii WU přiřazena pevná kreditová hodnota, která je předem vypočítána ze strany projektu. Přidělovaný kredit je momentálně (6/2007) nejvyšší ze všech projektů pod systémem BOINC.
Na závěr bych tedy zdůraznil, že na rozdíl od jiných biochemických projektů pod Boinc (Rosetta, Predictor, Protein, Tanpaku atd.), tento projekt počítá biochemické procesy od absolutního základu přírody jaký nyní známe -od kvantové mechaniky. To jej dělá výjimečným, protože kvantově mechanický popis je prozatím nejpřesnější, ostatní projekty využívají méně precizní a méně přesné metody,což ale neznamená, že jsou méně užitečné. Každý projekt otevírá svoje malé okno pro poznávání světa okolo nás, QMC se nedá v žádném případě použít na všechno.
Takže koho zajímá využití nejmodernějších výdobytků vědy pro popis života, může přispět k jeho rozvoji zapojením se do tohoto projektu na stránkách http://qah.uni-muenster.de.
Při přípravě článku bylo použito částečně překladu informací z webu projektu, z fóra projektu a další doplňující informace například od Dura, kterému bych za možnost jejich využití chtěl moc poděkovat. Za pomoc se závěrečnou korekcí bych chtěl poděkovat wertovi668 a za gramatickou a syntaktickou kontrolu JardoviM.
Svůj komentář na tento článek, co by mělo být opraveno, či doplněno můžete napsat do této sekce na našem týmovém fóru. Téma s komentářem k tomuto konkrétnímu článku, by mělo nést stejný název, jako článek na webu.

Napsal uživatel forest dne 13.06.2007 13:49