Hlavní menu
Sociální sítě:



Jedním ze základních cílů projektu Rosetta je předpovídat tvary, do kterých se proteiny rozvíjí v přírodě. Proteiny jsou lineární molekuly polymerů tvořených z monomerových aminokyselin; často se k nim odkazuje jako k řetězům (chain).
Napomůže analogie:Když máme kovový řetěz, může mít rozličné tvary podle toho, jaké síly jej namáhají. Například, pokud natáhneme jeho konce, řetěz se napne do rovné linie…a když jej pustíme na zem, dostane unikátní tvar. Na rozdíl od kovového řetězu, který je složen z identických článků, proteiny jsou tvořeny z 20 různých aminokyselin, kde má každá unikátní vlastnosti (například různé tvary, přitažlivé a odpudivé síly). Jejich kombinací a působením sil na řetěz vzniká specifický tvar, který nazýváme fold (ohyb, záhyb). Pořadí, ve kterém jsou aminokyseliny spojeny, je určující pro to, jak se bude protein ohýbat. Existuje mnoho druhů proteinů, které se liší počtem a pořadím jejich aminokyselin.
K určení toho, jaký tvar bude mít určitý protein v přírodě (o to se Rosetta snaží), je třeba nalézt fold s nejnižší energií. Energie se určuje několika faktory. Například, některé aminokyseliny jsou vzájemně přitahovány, takže jsou si v prostoru blízko; jejich interakce přináší žádaný energetický přínos. Strategie Rosetty k nalezení nízkoenergetických tvarů vypadá asi takto:
1. Začneme s plně otevřených řetězem (podobně jako s kovových řetězem napnutým za konce)
2. Posuneme s částí řetězce tak, abychom vytvořily nový tvar.
3. Vypočítáme energii nového tvaru.
4. Přijmeme nebo zamítneme posunutí podle změny v energii.
5. Opakujeme skoky 2 – 4 dokud každá část řetězu nebylo mnohokráte posunuta.
Tomu procesu říkáme trajektorie. Výsledkem trajektorie je předpovídaná struktura. Rosetta si pamatuje dráhu nízkoenergetického tvaru nalezené v každé trajektorii. Každá trajektorie je unikátní, protože posuny částí proteinu jsou zkoušeny podle náhodného čísla. Takže se vždy nenajde ta samá nejnižší energie, neboť existuje mnoho možností.
Trajektorie se může skládat ze dvou úseků. V prvním úseků je použita zjednodušená reprezentace aminokyselin, které umožňují rychle vyzkoušet mnoho tvarů. Mluvíme o hledání s nízkým rozlišením – na obrazovce uvidíte proteinový řetězec rychle poskakovat. V druhém úseku již Rosetta používá plnou reprezentaci aminokyselin. O této fází se mluví jako o uvolnění (relaxation). Místo toho, abychom s proteinem intenzivně hýbali, zkoušíme menší změny ve snaze posunout aminokyseliny do jejich správného uspořádání. Mluví se o hledání s vysokým rozlišením – na obrazovce uvidíte, jak se protein pomalu pohupuje. Rosetta je schopná projít prvním úsekem během pár minut na moderním počítači. Druhý krok trvá déle vzhledem ke zvýšené komplexitě plné reprezentace (všech atomů) aminokyselin.
Váš počítač bude typicky generovat 5-20 takových trajektorií (na jednotku) a poté projektu zpět zašle tvary s nejnižší energií každé trajektorie. Poté analyzujeme všechny nízkoenergetické tvary ze všech počítačů, abychom nalezli ty nejnižší. Ty se stanou předpovědí pro daný protein.
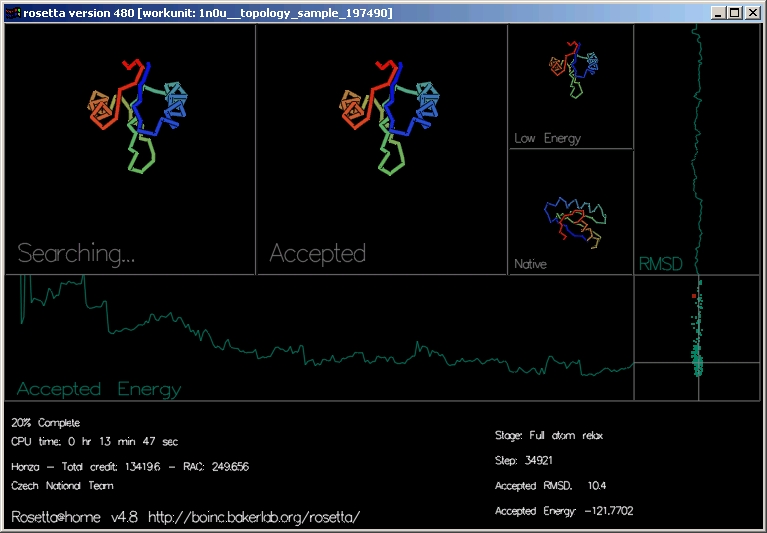
Grafika ukazuje průběh každé trajektorie a tvar proteinu ve 4 obdélnících.
"Searching..." (hledání) ukazuje pohyby, ze kterých Rosetta zkouší utvořit tvar. Můžete vidět tvar řetězu vybarvený barevnou duhou od modré do červené.
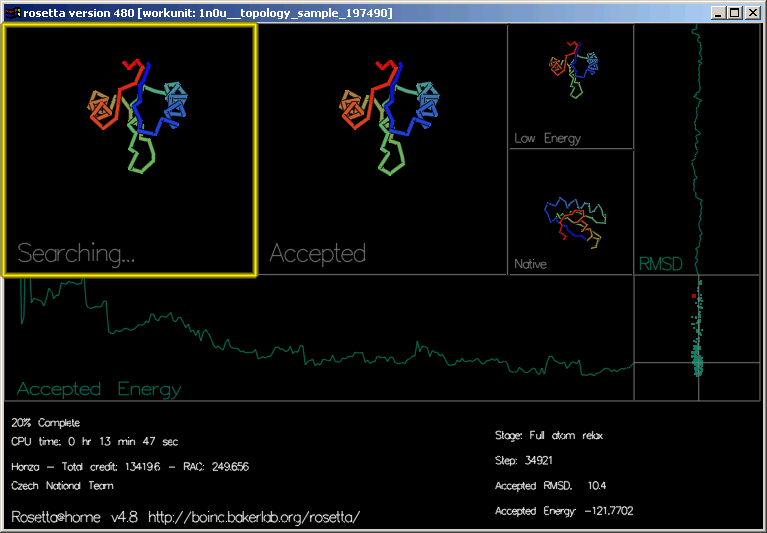
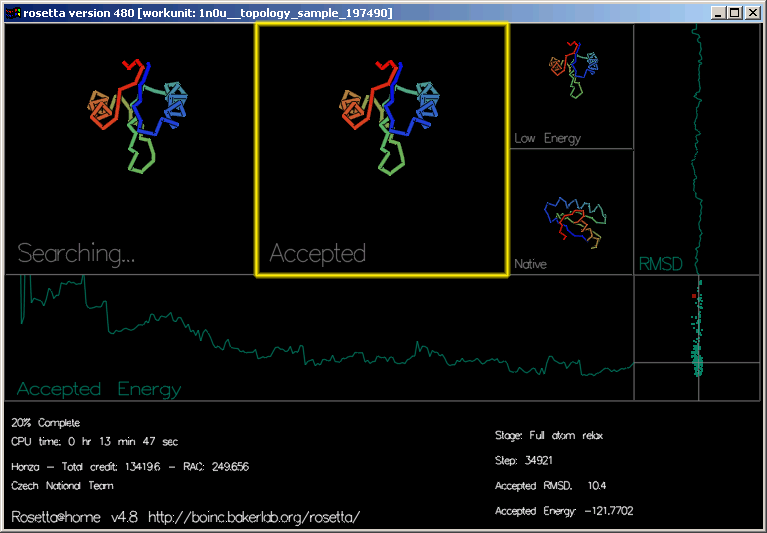
"Accepted" (přijatý) ukazuje naposledy přijatý pohyb.
"Low Energy" (nízkoenergetický) ukazuje tvar vykazující nejnižší energii v současné trajektorii.

"Native" (přírodní) ukazuje experimentálně určený skutečný tvar, pokud je známý.
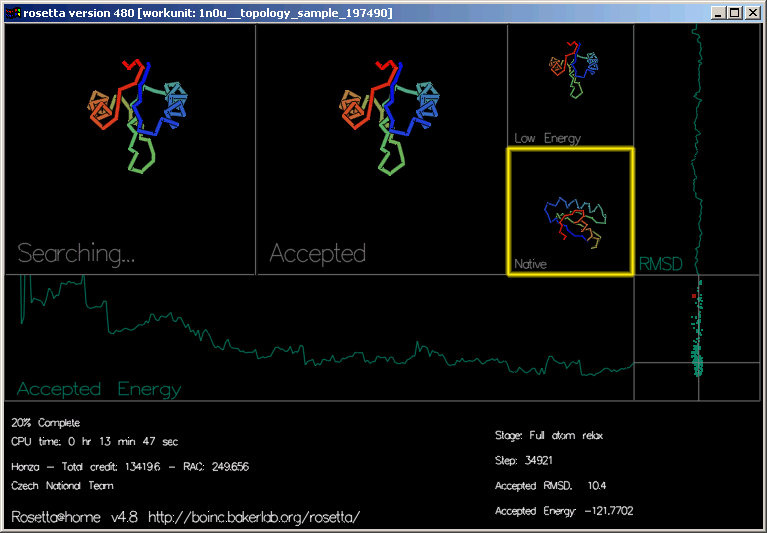
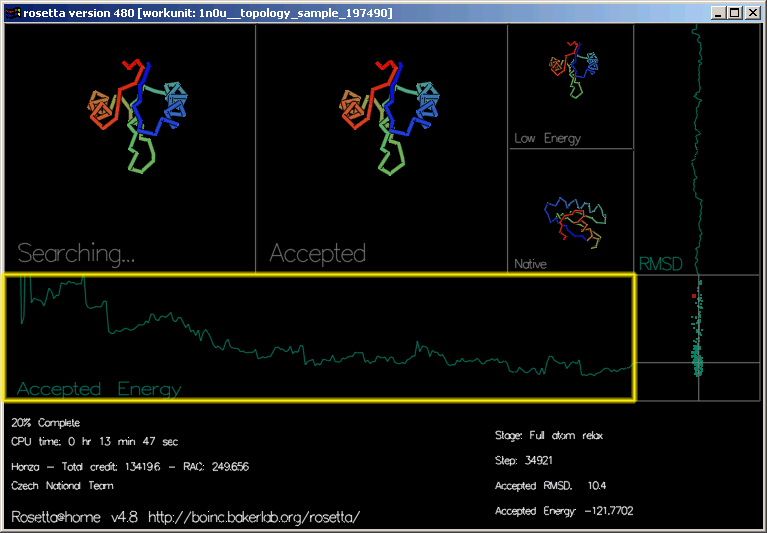
"Accepted Energy" (akceptovaná energie) je grafem ukazujícím energii každého akceptovaného pohybu této trajektorie. (osa x zobrazuje průběh trajektorie, osa y energii.)
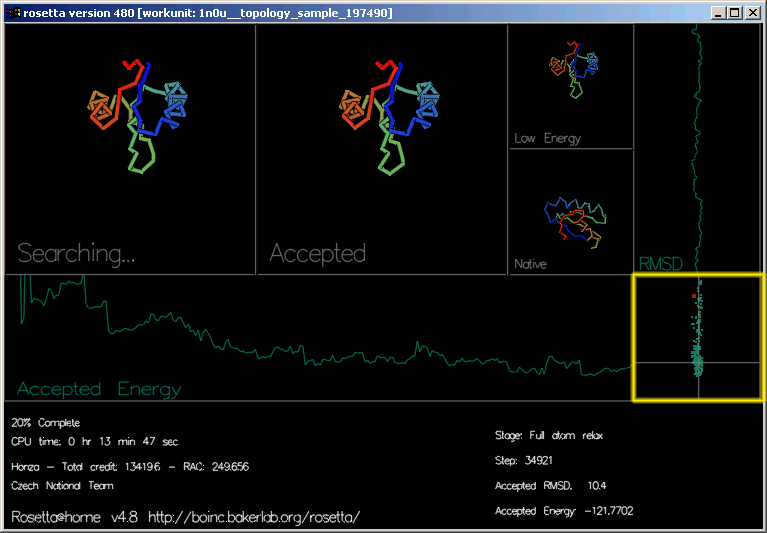
"RMSD" (relative mean square deviation; relativní odchylka vzdáleností čtverců) ukazuje, jak blízko je současná přijatá struktura správné odpovědi. (osa x je RMSD, osa y ukazuje průběh.)

Poslední obdélník vpravo dole vykresluje energii a RMSD jednotlivých akceptovaných pohybů. To jsou ty samé, které můžete vidět na stránce Top predictions (nejlepších předpovědí)
Avšak v grafice můžete vidět energii všech *akceptovaných* pohybů během jejich trajektorie. Vykreslení nejlepších předpovědí na webu je pouze z nízkoenergetických tvarů každé trajektorie (tedy ne všech akceptovaných)
P.S. Nejsem molekulární biolog či podobný odborník a v tématice se příliš nevyznám. Navíc neznám výrazy zavedené v našem jazyce, takže se omlouvám za novotvary a další prohřešky. Vycházel jsem z článku popisující dané téma, který jsem nahrubo přeložil + své poznatky s projektem, čtení fóra Rosetty atp. Za upozornění na případné chyby, nesrozumitelné pasáže či návrhy na zlepšení děkuji.
Honza
Zdroj: http://boinc.bakerlab.org/rah_graphics.php
Svůj komentář na tento článek, co by mělo být opraveno, či doplněno můžete napsat do této sekce na našem týmovém fóru. Téma s komentářem k tomu konkrétnímu článku, by mělo nést stejný název, jako článek na webu.