Hlavní menu
Sociální sítě:



Již několik let vědci porovnávají genové sekvence různých organismů, aby zjistili, zda mezi nimi existuje podobnost. Geneticky podobné sekvence mohou mít (a většinou i mají) totiž i podobnou funkci. Vědec který studuje genovou sekvenci nějakého organismu, může takto získat důležitou stopu o jeho úloze, pouze díky porovnání s podobnou genovou sekvencí známé funkce v jiném organismu.
Problém je v tom, že vědci objevují nové informace a zapisují je do mnoha různých databází, které obsahují informace o genových sekvencích. V průběhu let byla do záznamů v databázích proteinů připojena spíše velká část sekundárních informací (strukturální, funkční, podobnostní a různých křížových odkazů). Jakmile jsou tyto informace jednou zadány, zřídka kdy se aktualizují nebo opravují. Popis předpokládané funkce proteinu je proto často neúplný, nebo může být nesprávný. Odvození nové sekvence od předchozí nesprávné funkce, může přenášet tuto chybu na další nové záznamy. Navíc mnoho proteinů je složeno z několika strukturálních nebo funkčních domén (moduly obsahující odlišné evoluční, funkční a strukturní jednotky), které mohou být přehlédnuty automatickými anotacemi.
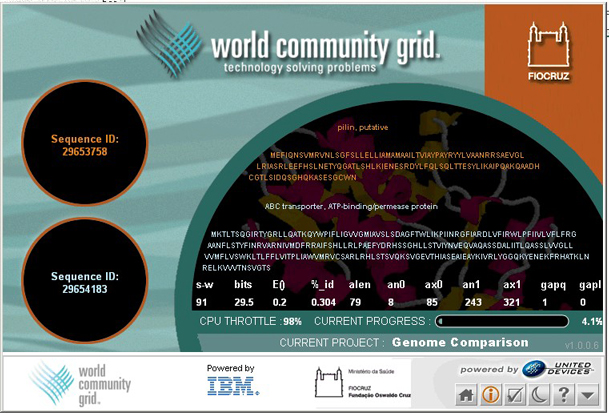
Hlavním cílem projektu Genome Comparison je poprvé provést kompletní párové srovnání všech známých proteinových sekvencí. Získané indexy podobnosti budou použity spolu se standardizovanou genetickou ontologií (www.geneontology.org/) jako referenční repozitář pro komunitu editorů. Ten bude následně poskytovat neocenitelný zdroj dat všem biologům. Program porovnání sekvenční podobnosti použitý v projektu Genome Comparison se nazývá SSEARCH. Využívá volně dostupného rigorózního algoritmu Smith-Waterman, který najde matematicky nejlepší lokální vztahy mezi dvojicemi sekvencí. Jelikož vědci zkoumají stále nové genomy z dalších organismů, mohou je zároveň přidat do této nově vznikající databáze, provést párové srovnání a přispět tak novými ověřenými daty dalším vědcům i samotnému vytřídění dat z databází od zbytečných chyb.
Výsledkem bude přesná anotace, korekce nesrovnalostí a přiřazení možných funkcí hypotetickým proteinům s neznámou funkcí. Navíc proteiny s více doménami a funkčními prvky budou správně vidět. Objeví se i vzdálené vztahy. Tím se zlepší kvalita a interpretace biologických dat a naše chápání biologických systémů, interakcí hostitel-patogen a interakcí s prostředím.
Zdroje: World Community Grid
Překlad: Dzordzik
Korektura: forest + JardaM

Napsal uživatel forest dne 23.03.2019 20:18